Building an enzymatic fuel cell.
November 7, 2019

I competed in iGEM this year, along with 5-10 other students (depending on how you count slackers and cheerleaders).
iGEM is a competition for high school, undergraduate and graduate teams. The competition has been running for the last 15 years, and this year it has over 300 teams competing, teams from all around the world. One of the main requirements to do well in the competition is to characterise new genetic parts and submit them to the ‘registry of standard biological parts’. Together the student teams of the past 15 years have built a vast library of parts that others can use.
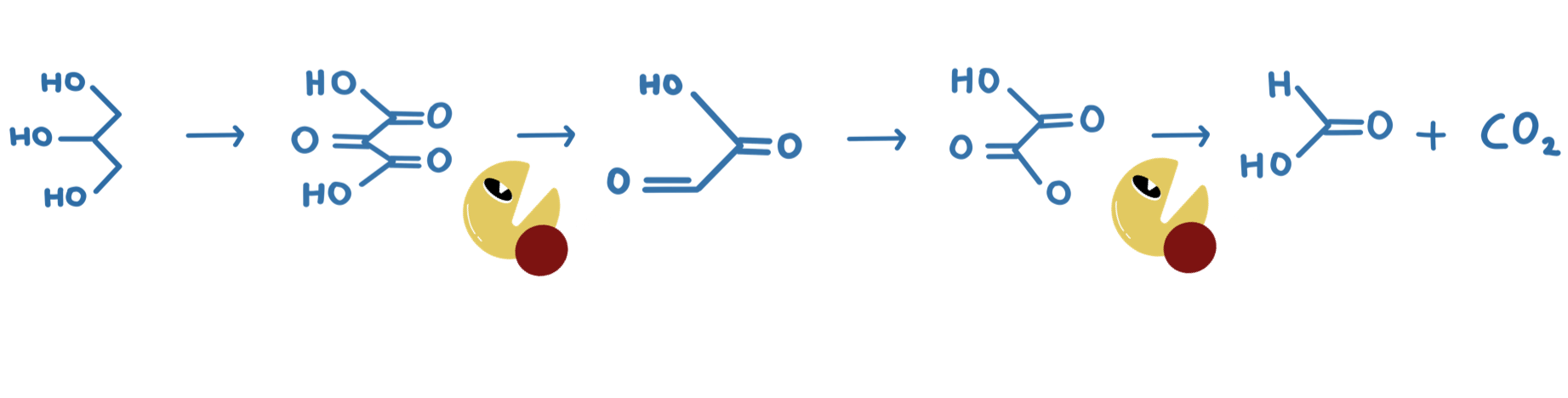
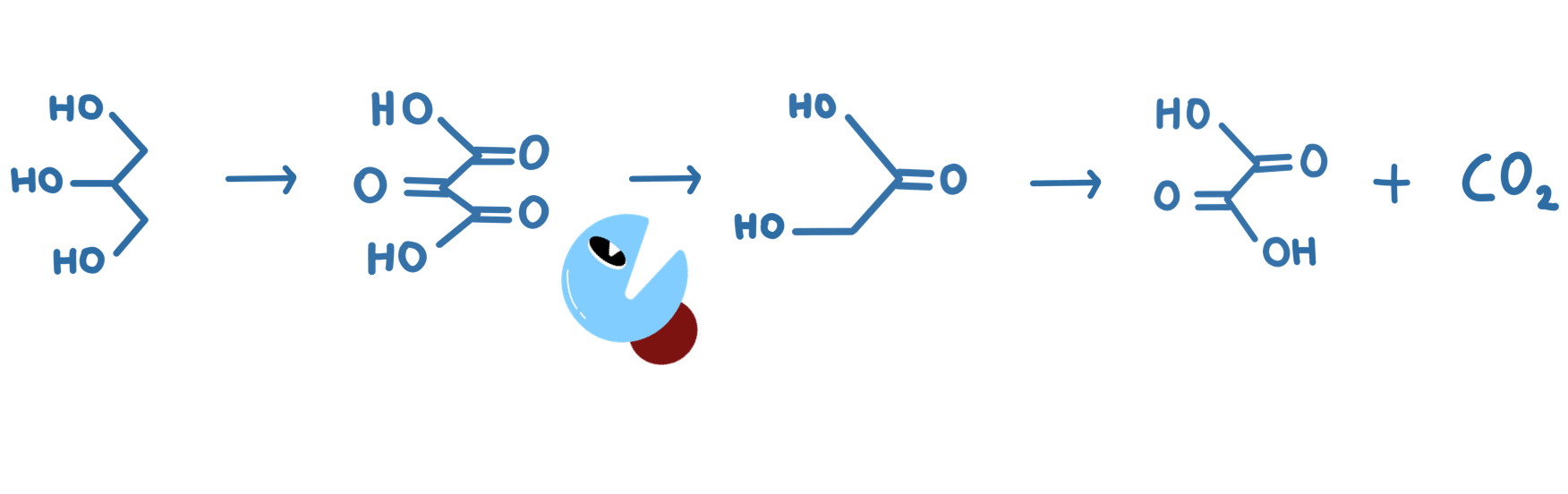
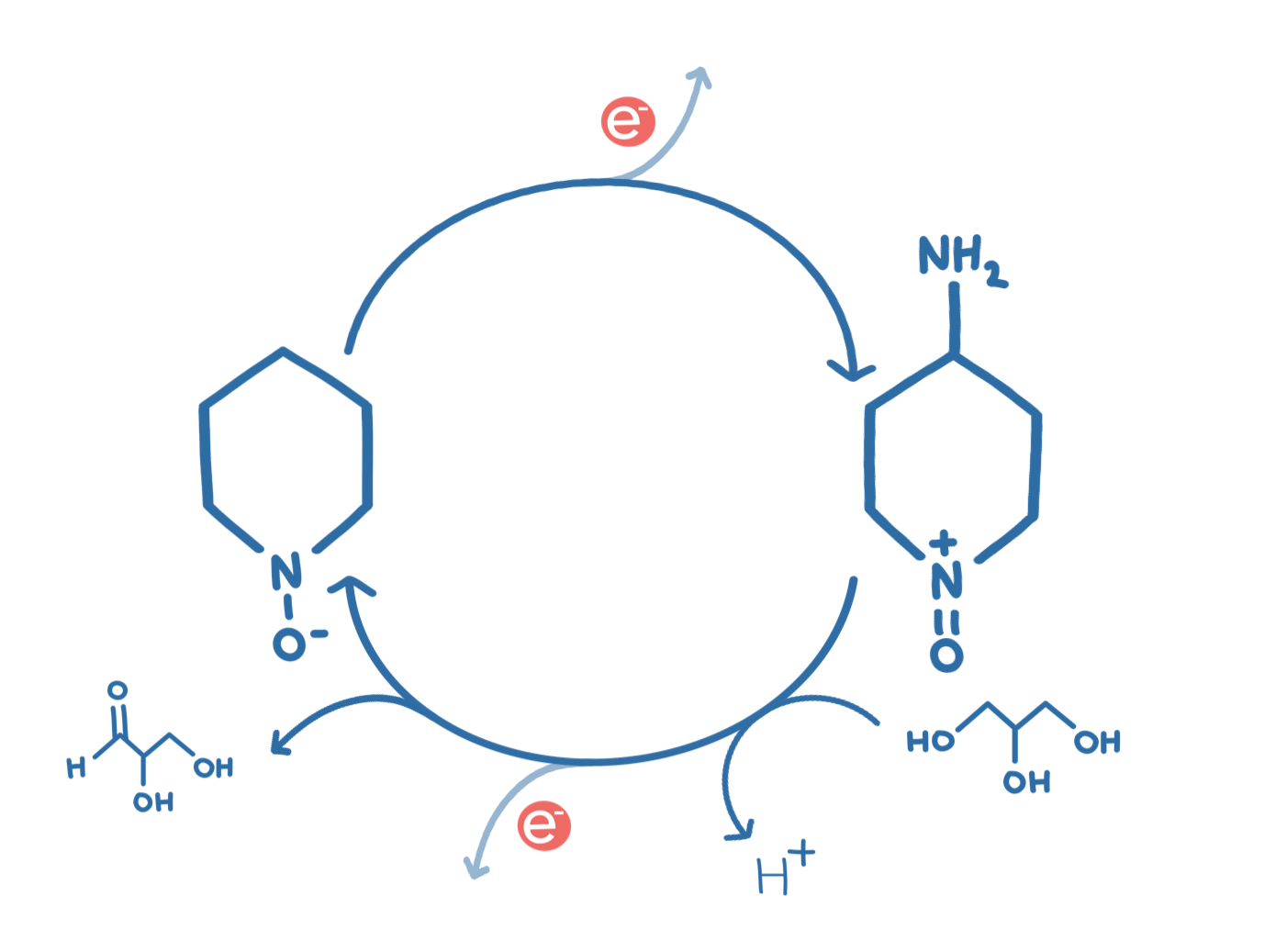
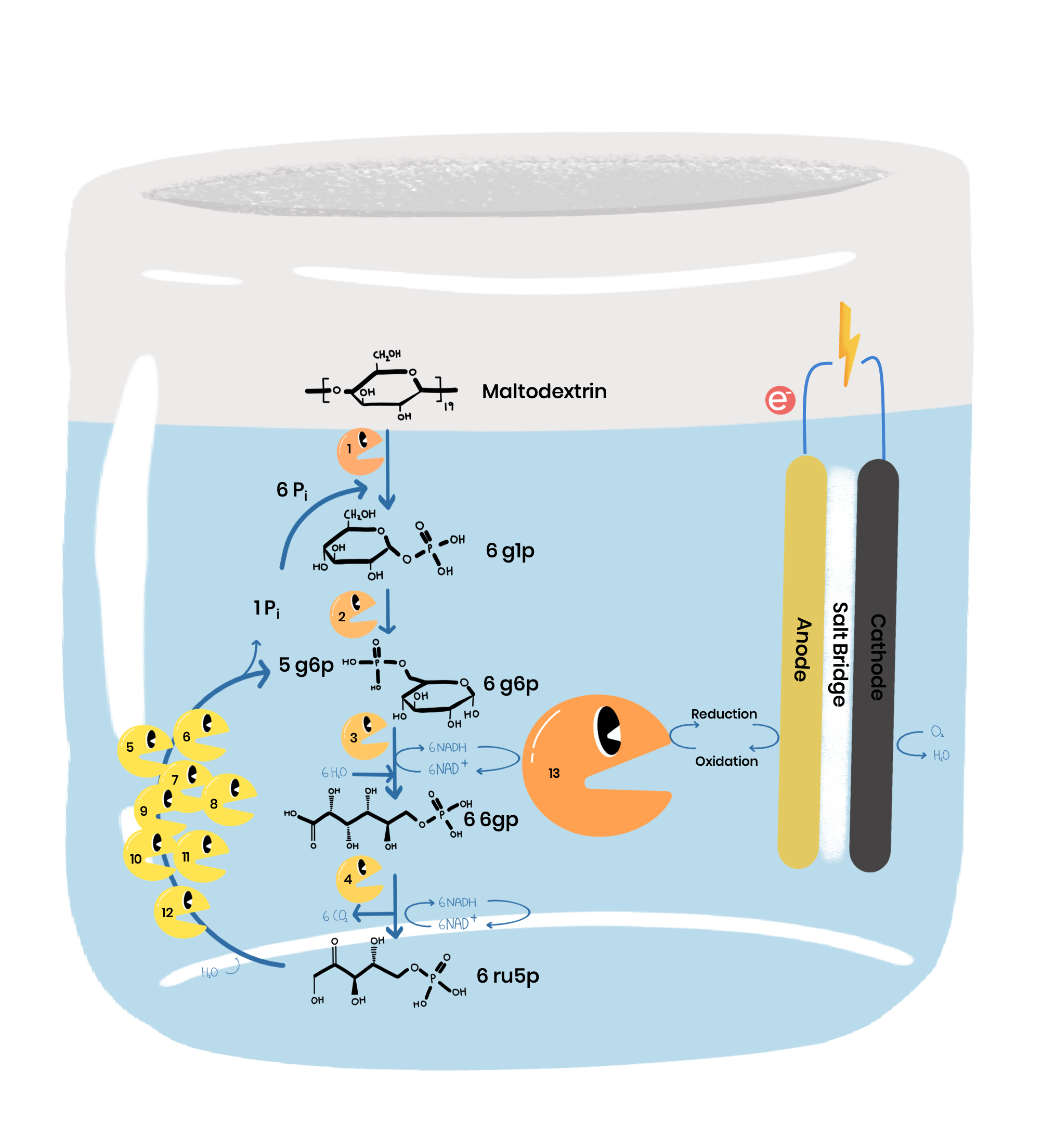
Our project was: “A glycerol based enzymatic fuel cell”.
![]()
An Enzymatic Fuel Cell (or EFC) is a battery-like device that exploits enzymes’ fantastic ability to catalyse organic reactions for electrical power. We are especially motivated by the idea of using enzymes to build greener energy sources and storage. Our contribution to this goal was to improve an existing glycerol fuel cell, where one of the enzymes used in the fuel cell naturally has a slow rate of reaction, a small pH range, and (like most enzymes) is not very stable at room temperature.
More details about our project can be found on our wiki.





Biochemistry
Ok ok, biochemsitry is a little more complex than I imagined. I live in the world of programming, where you have well defined types. Let 2 be an integer. It always behaves like an integer. It doesnt matter if I put an integer and a float into the same list. An integer is still an integer. In biochemsitry; chemicals interact when you don’t want them to, infections grow where they shouldn’t, genes mutate, the folding of an enzyme depends on its environment…
Everything reacts with everything else… How do you manage this kind of complexity?!





Me, in the lab!
After a 1 hour health and safety lecture, I was given access access to the VUW bio labs. In fact, my training in PC2 means I can supervise others in the lab… ??!? Me, with zero biological training. I could walk in and muck around with PCR machines, with live GM cultures, … But it turns out, the most dangerous things in the lab were the microwave and me.
Agar is a stubborn solid. To melt it you need to blast it in the microwave or around 10 mins. One time, we left the lid slightly too tight. That was scary. Another time I managed to spill super heated agar all over the microwave, the desk and the floor.
And me… I failed at making LB three times in my first week… First two times I read the instructions wrong, the next time I confused 0.2grams and 2 grams. Ok, so being inept isnt necessarily dangerous. But, it kinda is.
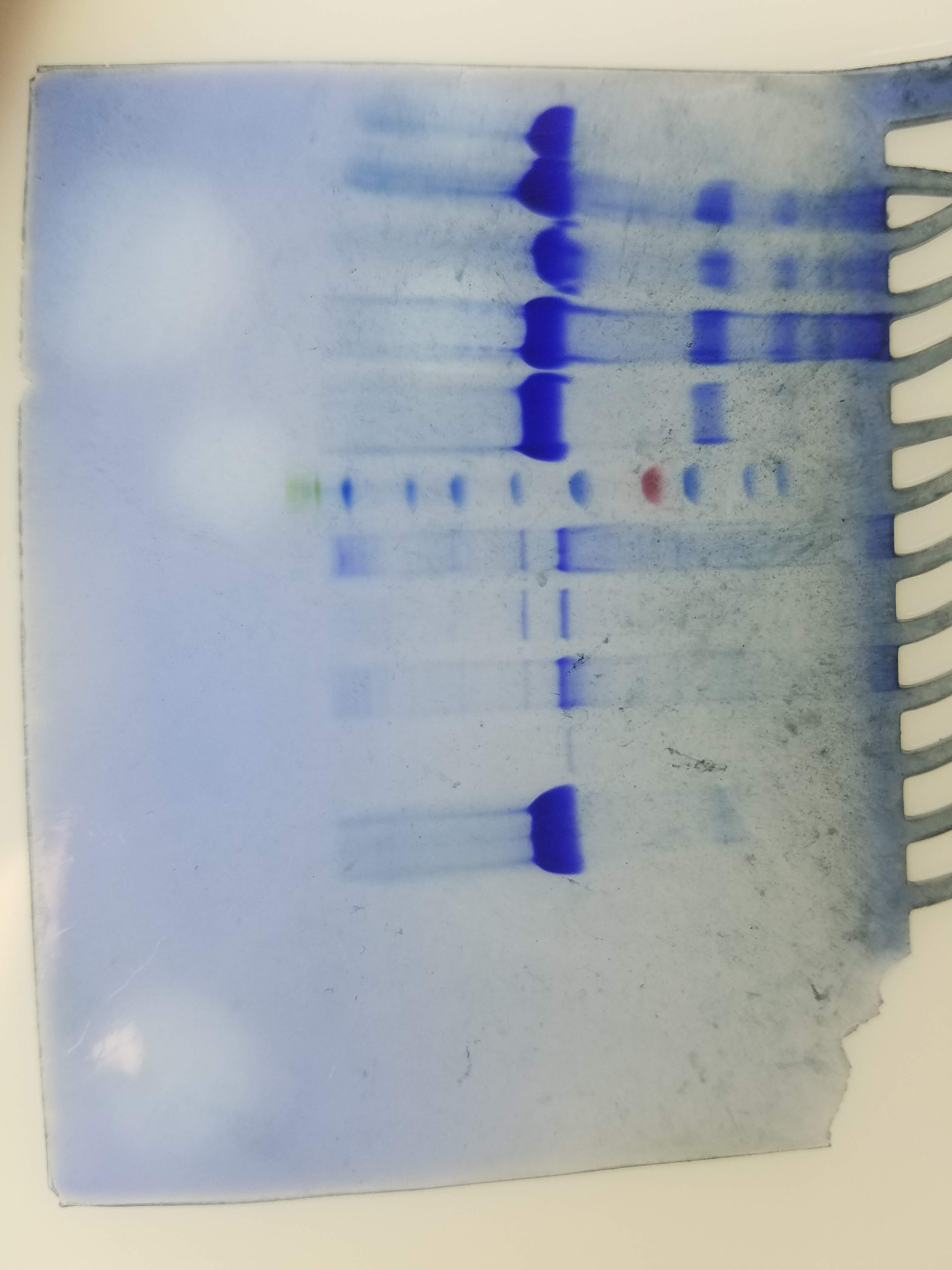
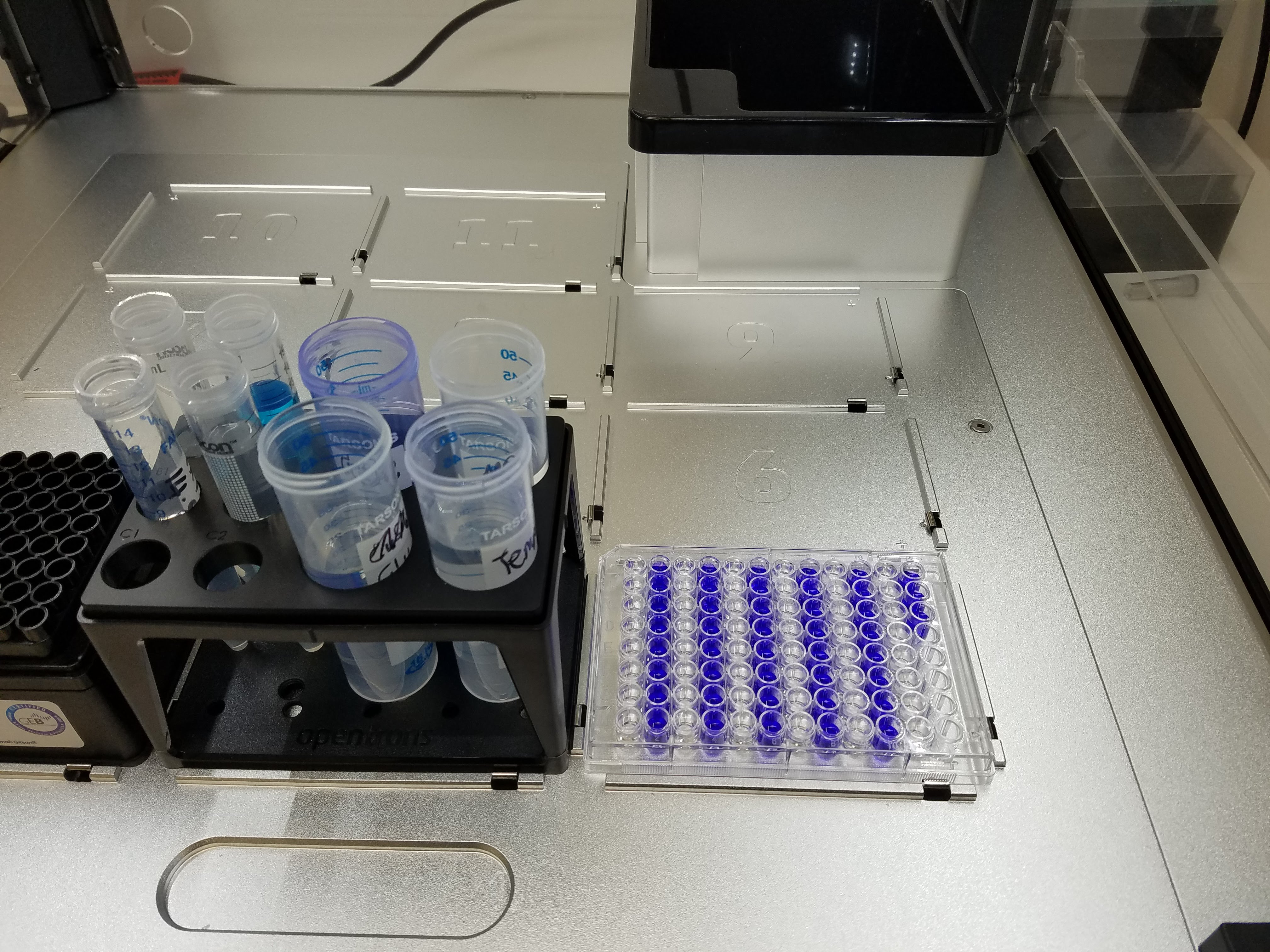


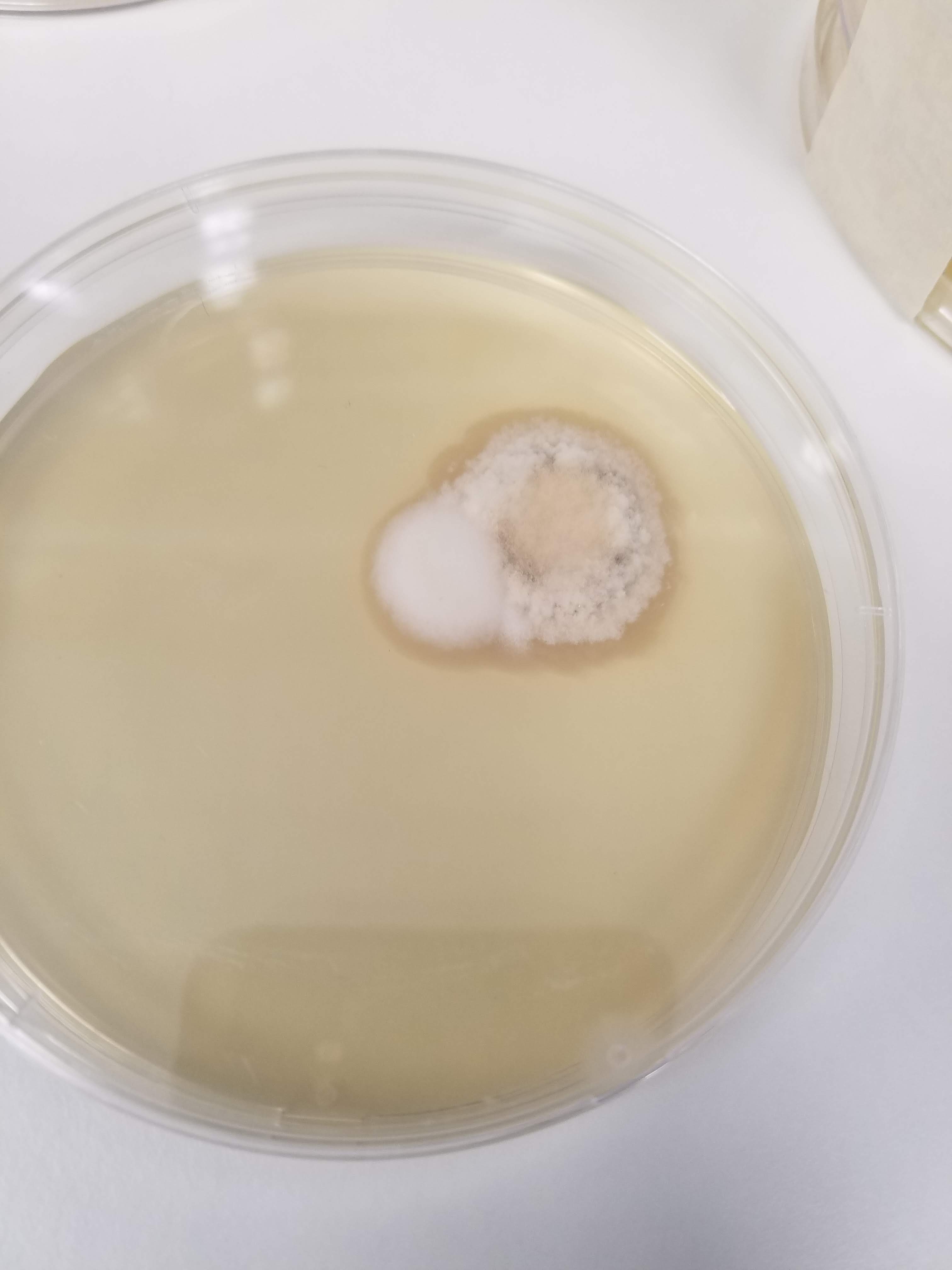
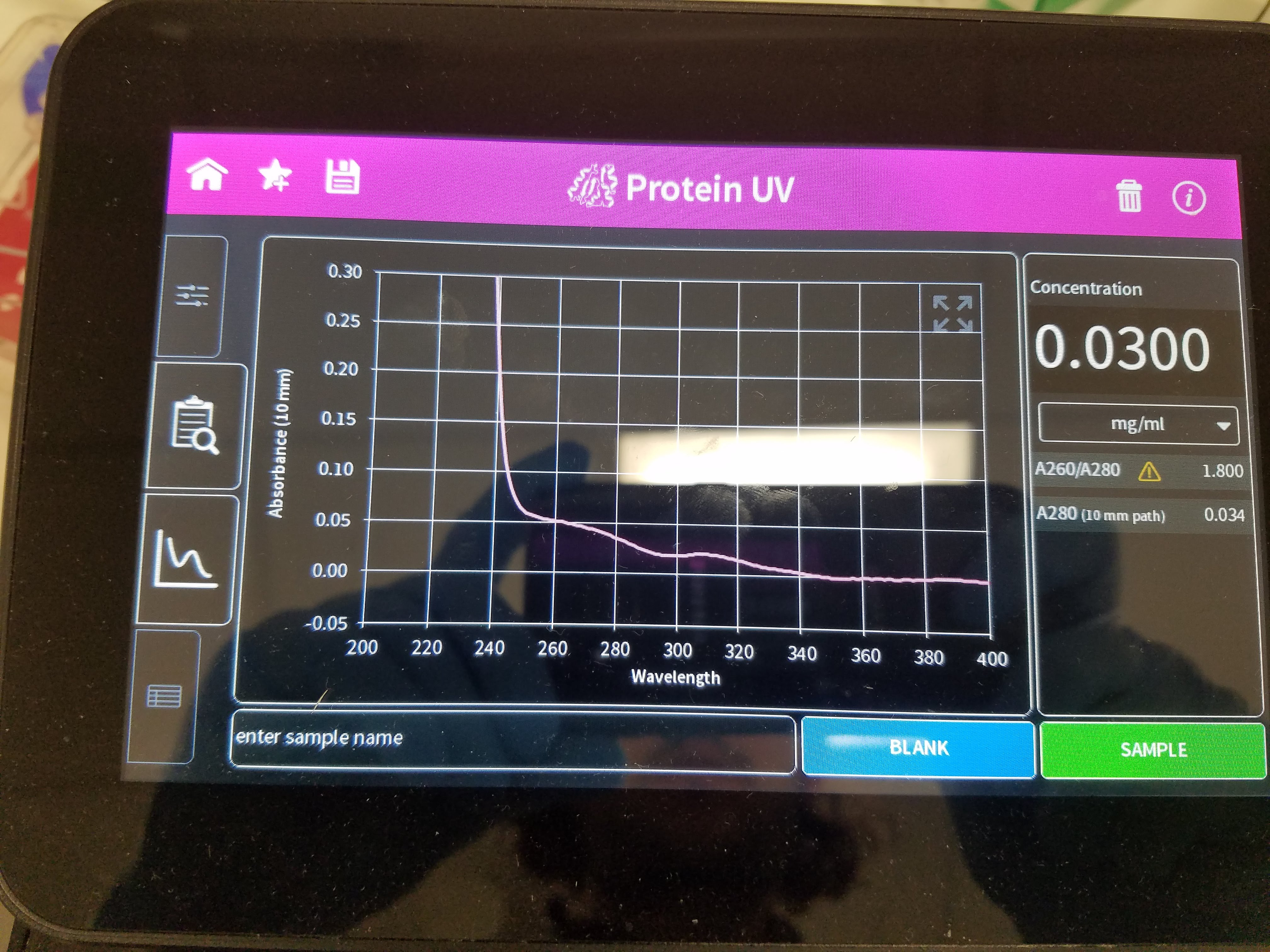
So, while doing the project, I did all sorts;
- sds page gel: separate protein based on size (using negatively charged particles - that bind to the protein - to drag it further based on it’s size)
- clean and concentrate: “push a solution containing protein or dna through a filter”
- transformation (via electroporation): electrocute some e. coli and hope that your chosen DNA / plasmid manages to slip through the e. coli’s perforated membrane
- digestion: cut a molecule into parts
- sonication: use a high frequency vibrator to create superheated bubbles, which shred a bacteria’s membrane (but somehow preserve the proteins)
- induction: add IPTG to our population of e. coli and this will activate the lac operon, thus telling the e coli to express our gene.
- plating: grow some bacteria on a plate of agar
- purification: sit in front of a very slow pump for a long time…
- his-tag purification: six histidines, which we added to our enzyme, bind to a nickel complex, which we placed in our filter. Our enzymes get stuck in the filter. Thus we will have separated them from everything else.
I found that a lot of the lab work felt like moving in the dark. One microlitre (a common amuont to be pipetting) is a really small amount, a twentieth of a drop. Effectivley, you are working with the invisible… You add one invisible drop of X and another invisible drop of Y and hope it works. You get very little visual feedback telling you have done the right thing. Much like being blind.






Building ontop of a large pile of nerd’s mistakes
Consider miniprep; a protocol for purifying plasmids from a culture of cells. This protocol consists of a series of simple steps; add reactant X, centrifuge for a minute, add reactant Y, centrifuge for a minute, … But, behind this simple protocol is a lot of ‘science’. And by ‘science’, I mean a lot of obsessed nerds and; their many bad ideas, their mistakes at 2am, and dissapointing dead ends.
How do we know that plasmids are soluble in certain cases while linear DNA is not? It is hard to imagine how that was discovered… (insert reference to the original research - pls help)
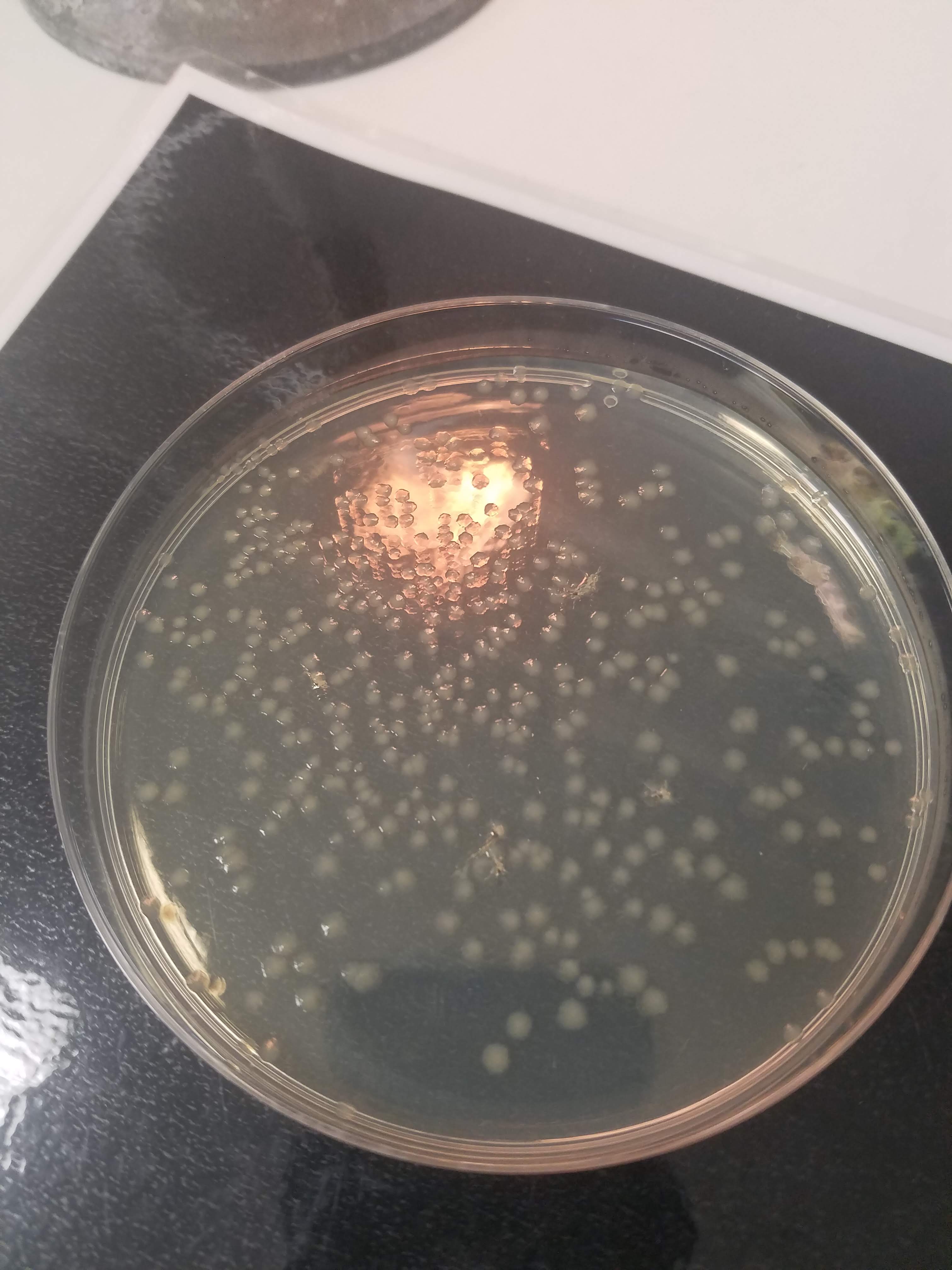



Mistakes and lessons
Here are some especially painful mistakes I made. Wince.
Be careful with the most expensive piece of equipment
The Sorvall was a large, expensive, centrifuge that could achieve accelerations around 50,000 times gravity. I was taught how to use it. Especially important was: the rotor (the thing that held the tubes) was expensive and made of carbon fiber. Because the Sorvall could generate such high accelerations, you needed to use special thicker test tubes.
I forgot about the specal thicker test tubes. Party via a miscommunication that confused things. But, simply, I forgot. The annoying thing was that there were a few little incroguities that I dismissed at the time, only to realise later that they were indicative of the problem.
We put some filter columns in the sorvall and set it going. Pop and bang. The filters sheared and crashed into the bottom of the tubes. We lost all our enzyme and I lost the trust of my advisors.
The puzzle of oscillating kinetics
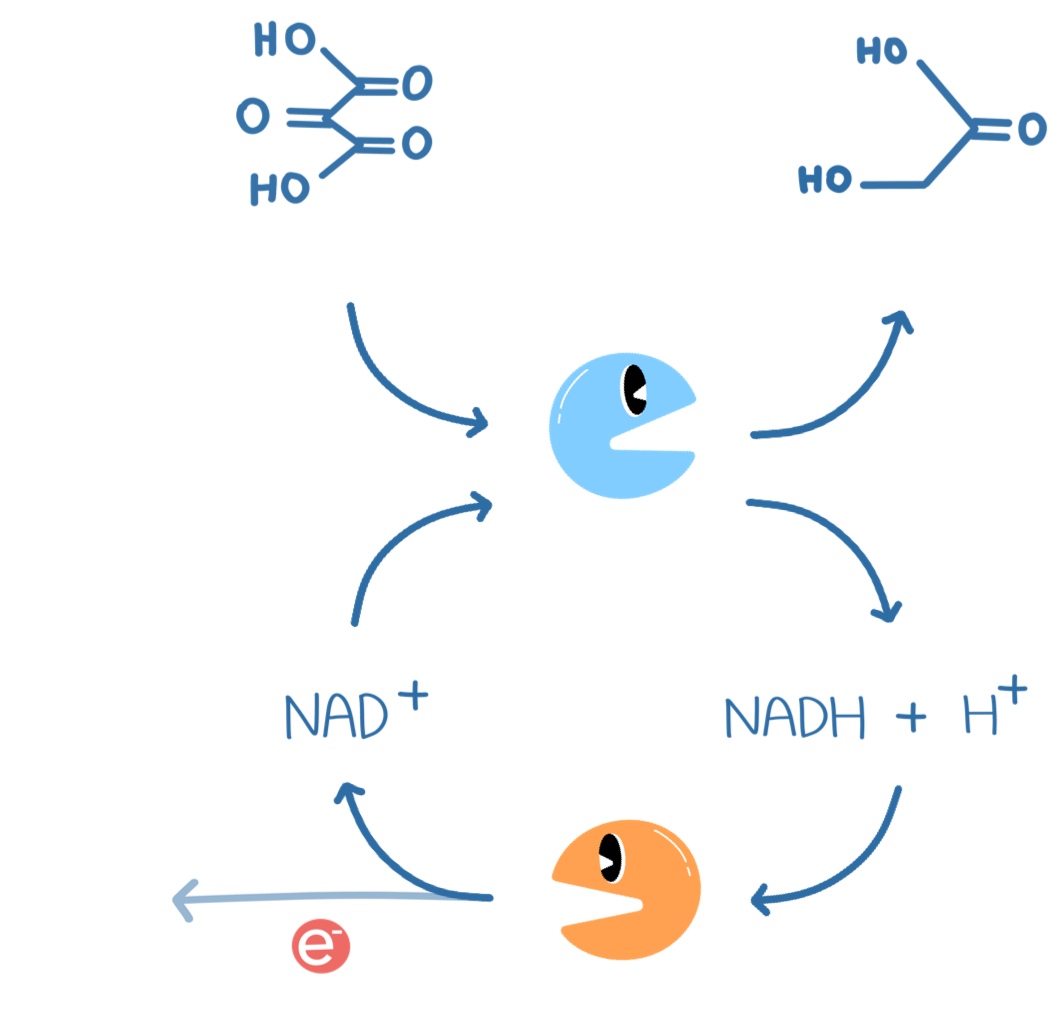
I was trying to measure the kinetics of formate dehydrogenase with mesoxalate. However, we were getting weird results that I couldnt explain. When just adding the enzyme to the buffer, I would see a steady increase in OD, then it would peak, finally, it would decay, slowly, back to the original OD (before the enzyme was added). These results seemed quite robust, I tried a few times.
My conclusion was that the enzyme and buffer were somehow reacting. I puzzled over this, but it did not make any sense to me.
What I should have done;
- questioned existing assumptions,
- checked the results of tests / controls I had already done,
- look at what we would expect to see theoretically.
After a long time puzzling, questioning others, and pulling my hair. Max retried a test I had already done; when adding the enzyme, give it a mix. Although, he gave it a really good mix, rather than my vauge jiggle.
Long story short. I learned that you need to mix the reaction…
Just order it from IDT
We needed to characterise an existing part for the iGEM registry. We could have ordered the part from the registry itself, instead we decided to order it via IDT. Part of our logic was that it would arrive sooner, also that we could use gibson assembly.
What we didnt plan on; I ordered the part with a slight, but very important error. Somehow (I have no idea how), I managed to add an extra base into the sequence, thus frame shiting the entire enzyme…
Because of this and some other (of my) mistakes, we couldnt get Silver. Eek!
Transformation. No, transformationsss
I attempted alot of transformations. I followed the protocol, it failed. I started again. It failed. I re-read the protocol, it failed. Hmph. I re-read the protocol, started again, it failed. FUCK! What am I doing wrong?
I am still not sure what I was doing wrong. The only difference I can see between my protocol, and a protocol that eventually worked (thanks to Alister, a postdoc), was that he incubated the digest and assembly for much longer than I did (5 mins vs an hour or more).




The Giant Jamboree in Boston
Ok, so the Giant Jamboree was awesome. There was science and there were world changing projects. Of the presentations I saw, my favourites were;




![]()

- TUDelft: Orthogonal replication
- NUS: Energy saving chassis
- Stanford: In vitro evolution
- BrownStanfordPrinceton: Astro pharmachy
- EPFL: Detection of flavescence dorée
- Great bay SZ: Dyed spider silk
- DTU-Denmark: Artificial promoter library
- Freiburg: Sterio isomer proteins
It was announced that the 2021 iGEM will be in Paris. Cool.
And that the ultimate goals of the competition are changing: iGEM will solve the SDGs. Awesome! To be honest, this announcement nearly made me cry. It is just so beautiful to me. I can see the potential: motivated people, working together, using powerful tools to help others.
Return of investment
My teams contribution was small in technical content, but we can now see how a glycerol fuel cell might work at an industrial scale, turning waste from biodiesil production into electricity. Many other teams had a similar lack in technical contribution. Like us they encountered misguided protocols and stubborn bacteria. But there were also tens of teams that made sizeable contributions for million or even billion dollar industries.
I was amazed by how much was achieved by small groups of students working part time. But, this amazement also made me sad. Look how much can be done when the ‘right’ projects are funded. Our society only spends ~$20 million (360 teams x $60,000) per year on iGEM teams, but billions on other things (don’t get me started…).
Motivated for next time
We, Victoria_Wellington, didn’t do very well. We were not awarded bronze because we placed some of our results on the wrong webpage. And without bronze, we couldn’t get silver, sigh… Next time!
Next time we are going to be better. Some ideas:
- Use onepot to make transposeases for meta genomic libraries
- What other molecules can be spider silk. What other properties could we get? Conductivity, ?
- Is it possibile to easily diagnosis DHA defficiency? We could then efficiently manufacture a supplement!
- Construct biological versions of electrical components; clocks, flip-flops, magnetic field sensors, capacitors, accelerometer, floats, …
- Most bacteria cannot be cultured in the lab. Use orthogonal expression to make these bacteria culturable in the lab!
- Can we program small multi-cellular organisims to sort waste / micro pastic?
- Simulate the interaction between antibodies and synthetic milk proteins to find anti-allergenic milk proteins.
Solving problems
A main theme amongst the nominees for best project, three out of six was: local problems, local solutions. Calgary, EPFL, Wageningen. All solved problems for local farmers of canola oil, grapes, olives. These farmers were struggling controlling various diseases or effects of the environment. These issues are the “difference between profit and loss” for local farmers.
What are local problems in NZ?
This question is actually a lot harder to answer than I thought it would be.
I am now more motivated to solve some applied problems. To find someone who is struggling with something, and help them.